-
Intergrative Genomics View (IGV) 사용법genome data analysis/Genome analysis 2020. 5. 5. 16:35
Intergrative Genomics View (IGV)는 가지고 있는 염기서열(bam file 이용)을 시각화해서 보여주고 보유한 유전체 염기서열의 depth 도 확인 가능합니다. 이 프로그램은 윈도에서도 사용 가능합니다.
1. IGV 프로그램 Download 및 설치 (2.8.0 버전 이용)
https://software.broadinstitute.org/software/igv/download
Downloads | Integrative Genomics Viewer
Did you know that there is also an IGV web application that runs only in a web browser, does not use Java, and requires no downloads? See https://igv.org/app. Click on the Help link in the app for more information about using IGV-Web. Install IGV 2.8.2 See
software.broadinstitute.org
2. 참조염기서열, bam, bai 파일 준비
- 참조 염기서열 파일 (reference sequence file) : fasta 파일만 있어도 되지만 유전자 이름 파일 등 IGV에서 요구하는 파일이 더 있으면 더 visual 하게 볼 수 있습니다. IGV에 등록된 ref 파일을 사용할 경우는 필요 없음)
- input file : IGV에서 보고자 하는 sample의 bam file
* bam file은 같은 fastq 파일을 이용해 만들었더라도 참조 염기서열을 어떤 것으로 썼냐에 따라 다른 파일이 됩니다. 최신의 참조 염기서열을 이용해 bam file을 만드는 것이 좋습니다. 최신의 참조 염기서열은 기존에 누락된 데이터들이 포함되어있는 경우가 많아, 이를 이용할 경우 더 많고 더 정확한 정보를 가진 bam file을 만들 수 있습니다. 아래 글처럼 참조 염기서열은 계속 업데이트되고, 특히 Pacbio 이용 염기서열 데이터 생산을 통해 예전보다 훨씬 더 정확한 참조 염기서열이 계속 만들어지고 있습니다. 그래서 Pacbio를 이용해 새롭게 샘플의 염기서열 해독을 진행하거나, 기존에 보유하고 있던 fastq파일을 새롭게 만들어진 참조 염기서열을 이용해 mapping 해서 쓰면 훨씬 더 좋은 데이터를, bam 파일을 만들 수 있습니다(새롭게 업데이트 안 된 참조 염기서열은 제외).
<참조 염기서열 업데이트 예시>
dongascience.donga.com
<VGP 프로젝트>
vertebrategenomesproject.org
그리고 항상 언제 참조 염기서열을 다운로드하였고, 어떤 이름의 reference 염기서열을 썼는지 기록해두는 게 좋습니다. 그래야 논문을 쓸 때, 다른 파일들과 비교할 때 정확하고 유용하게 사용할 수 있습니다.
3. genome file 만들기
만일 미리 갖고 있거나 IGV 내 .genome 파일 리스트에 있는 것이면 안 만들어도 됩니다. 리스트에 있는 것들은 여러 “부가적인 것들” (유전자 위치, 유전자 이름 등이 포함되어 IGV를 통해 좀 더 자세한 정보들 (유전자 이름, 위치 등)을 볼 수 있습니다. 리스트에 없으면 만들어야 합니다. IGV 프로그램 맨 위쪽 도구 창에서 genomes를 클릭한 후 create. genome file을 클릭합니다. Unique identifier와 Descriptive name에는 구분할 수 있는 이름을 넣어주고 FASTA file 항목에 reference.fasta (레퍼런스 시퀀스) 파일을 넣어줍니다.

왼쪽 위 "hg19"는 .genome file이 생성되면 자동으로 생성된 genome 파일로 변경됩니다. 참조 염기서열 파일로 .genome 파일을 만들면 fai 파일 (fasta 인덱스 파일)라는 파일이 생성됩니다. 그래서 일부러 fai 파일을 안 만들어줘도 됩니다만 참조 염기서열 파일 내용을 수정했다면 기존 fai 파일을 없애야 합니다. 만일 fai 파일을 삭제하지 않고 reference 파일 내용을 수정만 하면 수정내역이 반영이 안 됩니다.
4. bai file (bam index file)만들기
bai 파일을 만들기 위해서는 bam 파일이 필요한데 혹시 이게 필요하다면 아래를 먼저 보세요.
2020/04/04 - [genome data analysis/Genome analysis] - Fastq to consensus sequences
bai 파일은
samtools index sample.bam
이라고 하면 bai 파일이 만들어집니다. 이때 index 파일이 안 만들어질 수 있습니다. Bam file이 sorting이 되어있지 않다면 아래와 같은 과정을 진행해야 합니다. 이것도 "3"에 링크된 곳에 설명이 있습니다. 간략하게, 아래와 같이 하면 됩니다.
samtools sort sample.bam > sample.sorted.bam
5. bam 파일 어떻게 생겼는지 보기
Bam 파일은 binary 파일이라
less bam 파일이름
이렇게 해도 어떻게든 열 수는 있으나 봐도 무슨 말인지 모릅니다. 어떤 형식으로 생겼는지 파일을 보기 위해서는
samtools view x.bam | less
이렇게 쳐야 합니다. View 명령어가 왜 중요하냐면 IGV에서 이름들을 통일해야 할 때가 있어서입니다. Reference 파일 안에 있는 염색체 이름과 bam 파일 내의 염색체 이름이 같아야 합니다. 예를 들어 참조 염기서열 파일이 " > chr01"로 시작하는데 bam 파일 시작은 "> chr1...으로 시작한다면 IGV 실행 시 오류가 생깁니다. Bam file은 무거워서 수정이 힘들 수 있으니 이럴 경우 referece 파일의 시작 부분을 bam file의 시작 부분에 맞춰주면 됩니다.
아래는 오류 예시입니다.
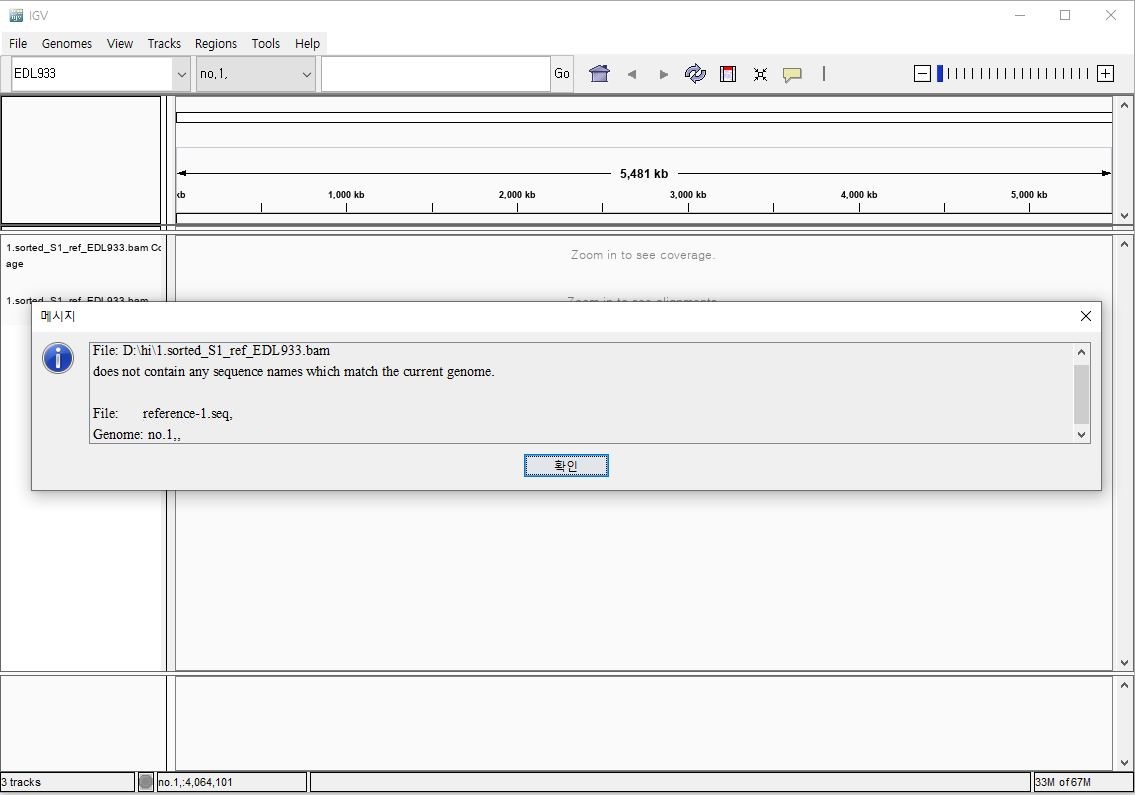
. genome 파일은 no.1....으로 시작하는데, bam과 bai 파일은 reference...로 시작하기 때문에 매칭이 안되고 있습니다. 이때 먼저 해야 할 것은
a. 기존 생성. genome 파일, fai 파일을 삭제한다
b. 참조 염기서열을 연다.
c. 참조 염기서열의 첫 행을 지우고 bam 파일의 첫행을 넣는다(여기서는 reference....)
d. 참조 염기서열을 새로 저장하고 다시 .genome 파일을 만든다
6. bam 파일 입력하기
도구 창에 File > Load from file… 을 클릭한 후 bam 파일을 입력합니다. 그러면 아래와 같은 그림을 볼 수 있습니다. 처음만 어렵고 다음번에는 참조 염기서열 준비, bam file 준비 후 bai 파일 뚝딱 만들고 IGV 안에 입력하면 돼서 별로 시간이 걸리지 않습니다.


프로그램 오른쪽 윗부분(빨간 동그라미) 부분을 수정하면 좀더 넓은지역을, 좁은지역을 변경해가면서 coverage, 염기서열을 확인할 수 있습니다. SMALL'genome data analysis > Genome analysis' 카테고리의 다른 글
Insilico PCR including Batch option (0) 2021.09.12 awk 다루기 - Data preprocessing (0) 2021.03.01 Multiple genome alignment, Synteny map 그리기 - Mauve (0) 2020.04.18 Variants Call Format (VCF) 파일 하나로 합치기 (0) 2020.04.17 Local BLAST - makeblastdb 이용 nhr, nin, nsq 파일 만들기 (0) 2020.04.15